分子建模

分子建模包含所有方法,即理論和計算,用於模擬或模仿分子的行為。這些方法用於計算化學,藥物設計,計算生物學和材料科學領域,用於研究從小型化學系統到大型生物分子和材料組件的分子系統。最簡單的計算可以手工執行,但是不可避免地需要計算機來對任何合理尺寸的系統進行分子建模。分子建模方法的共同特徵是分子系統的原子水平描述。這可能包括將原子視為最小的單個單位(分子力學方法),或用其光子(一種量子化學方法)將其用夸克,抗夸克和膠子和電子對質子和中子進行顯式建模。
分子力學
分子力學是分子建模的一個方面,因為它涉及使用經典力學(牛頓力學)來描述模型背後的物理基礎。分子模型通常將原子(核和電子共同描述為具有相關質量的點電荷)。相鄰原子之間的相互作用是通過春季樣相互作用(代表化學鍵)和范德華力來描述的。 Lennard-Jones潛力通常用於描述後者。靜電相互作用是根據庫侖定律計算的。原子是在笛卡爾空間或內部坐標中分配的坐標,也可以在動態模擬中分配速度。原子速度與系統溫度有關,即宏觀數量。集體數學表達稱為潛在函數,與系統內部能(U)有關,該系統的熱力學數量等於電勢和動能的總和。最小化勢能的方法稱為能量最小化方法(例如,最陡峭的下降和偶聯梯度),而對系統的行為進行建模隨時間傳播的方法稱為分子動力學。


該函數稱為潛在函數,將分子勢能計算為一個能量術語的總和,描述了鍵長,鍵角和扭轉角度遠離平衡值的偏差,以及描述非原子的術語的術語der waals和靜電相互作用。由平衡鍵長,鍵角,部分電荷值,力常數和范德華參數組成的一組參數集體稱為力場。分子力學的不同實現使用不同的數學表達式和不同的參數來實現潛在功能。當今使用的公共力場是通過使用化學理論,實驗參考數據和高量子計算而開發的。該方法(稱為能量最小化)用於查找所有原子的零梯度位置,換句話說,是局部能量的最小值。低能狀態更穩定,並且由於其在化學和生物過程中的作用而經常進行研究。另一方面,分子動力學模擬計算系統的行為隨時間的函數。它涉及解決牛頓的運動定律,主要是第二定律, 
變量
分子可以在真空中建模,也可以在溶劑(例如水)的存在下進行建模。真空中系統的模擬稱為氣相模擬,而包括溶劑分子的存在的模擬稱為顯式溶劑模擬。在另一種類型的模擬中,使用經驗數學表達來估計溶劑的效果。這些稱為隱式溶劑化模擬。
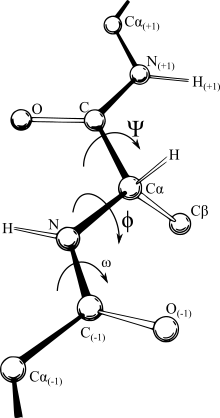
坐標表示
大多數力場都依賴距離,這是這些笛卡爾坐標最方便的表達。然而,特定原子之間發生的鍵的相對剛性性質,本質上定義了名稱分子的含義,使內部坐標系成為最合乎邏輯的表示。在某些字段中,IC表示(鍵長,鍵之間的角度和鍵的扭角)稱為z-matrix或扭轉角表示。不幸的是,笛卡爾空間中的連續運動通常需要內部坐標中的不連續的角分支,這使得與內部坐標表示中的力場相對困難,而相反,在笛卡爾空間中,原子的簡單位移可能不是直線軌跡互連鍵的禁令。因此,計算優化程序在其迭代過程中在表示之間來回翻轉非常普遍。這可以主導電勢本身的計算時間,而在長鏈分子中會引入累積數值不准確性。儘管所有轉換算法都會產生數學上相同的結果,但它們的速度和數值準確性也有所不同。當前,對笛卡爾轉換最快,最準確的扭轉是自然的擴展參考框架(NERF)方法。
申請
分子建模方法通常用於研究無機,生物學和聚合系統系統的結構,動力學,表面特性和熱力學。如今,數據庫中很容易獲得大量的力場分子模型。已經使用分子建模研究的生物學活性類型包括蛋白質折疊,酶催化,蛋白質穩定性,與生物分子功能相關的構象變化以及蛋白質, DNA和膜複合物的分子識別。